
This blog is about conformity assessment procedures under the MDR. Conformity assessment means the process demonstrating whether the requirements of the regulation relating to a device have been fulfilled. Like the MDD, the MDR allows you to choose between different procedures to CE mark your medical device depending on the risk class of the device. Medical devices can fall into the following risk classes: I (lowest risk), Is (sterile), Im (with measuring function), Ir (reusable surgical instrument), IIa, IIb and III (highest risk).
Key changes in conformity assessment procedures in the MDR compared to the MDD:
- Full quality assurance system (MDD Annex II – MDR Annex IX):
Increased number of Technical Documentations to be assessed by the Notified Body (NB) - EC type-examination + product verification (MDD Annex III + Annex IV – MDR Annex X + Annex XI, Part B):
Every product needs to be tested (no sampling) - EC type-examination + production quality assurance (MDD Annex III + Annex V – MDR Annex X + Annex XI, Part A): not for class III devices any more
- EC type-examination + product quality assurance (MDD Annex III + Annex VI): deleted
- New pre-market scrutiny for high-risk devices
For medical devices that fall in class I, as a manufacturer you can declare the conformity and affix the CE-mark as described in Annexes IV and V under your own responsibility without involving a notified body. For all other devices you have to involve a notified body in the conformity assessment.
For some high-risk devices, specific procedures – some including scrutiny by an expert panel – have to be applied:
- class III implantable devices (Annex VIII, rule 8) and class IIb devices intended to administer medicinal products (Annex VIII, rule 12)
The expert panel can judge based on a clinical evaluation assessment report prepared by the notified body whether the clinical evidence provided by the manufacturer is sufficient to provide confidence in the safety and performance of the device and whether an additional scientific opinion is required. - devices incorporating medicinal products (Annex VIII, rule 14)
Apply Annex I of the medicinal products directive 2001/83/EC. NB shall consult medicinal products authority. - devices manufactured utilizing tissue from human or animal origin (non-viable) (Annex VIII, rule 18)
Respective directives and regulations must be applied and competent authorities consulted. - devices composed of substances (Annex VIII, rule 21)
Apply Annex I of the medicinal products directive 2001/83/EC. NB shall consult medicinal products authority.
The following flowchart shows the different procedures that can be used depending on the risk class of the device.
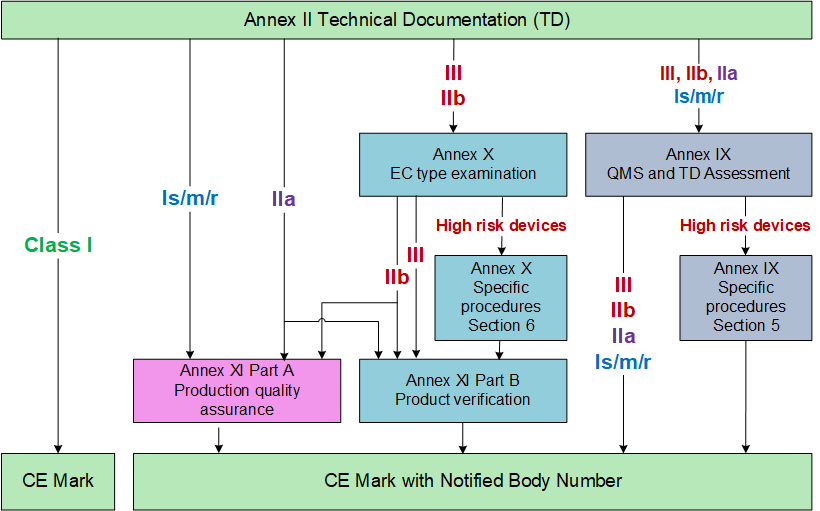
The slides for you to download are intended to provide you with more details of the conformity assessment procedures. The presentation does not purport to be complete and I also recommend to keep track of ongoing discussions and monitoring of publications.
I just can recommend to talk to your notified body as soon as possible to discuss with him certification of your products under the MDR. If you newly need a notified body, because your device was class I under the MDD but now is class Ir (reusable surgical instrument), class IIa, IIb or III (e.g. some products incorporating substances) under the MDR, it is even more important to talk to a notified body to have a chance to get your device certified before the end of the transition period in May 2020.
If you are unsure about the classification of your device and the selection of the conformity assessment procedure that suits best the needs of your company, get in contact with us to take advantage of the experience we are constantly able to expand through our work.
by Dr. Sabine Nieba, Senior Consultant, confinis ag